Sequencing of Repeat Expansions in Molecular Diagnostics: Comparing Hybridization-based and CRISPR-Cas9-based Approaches
October 7, 2025
Bettina Vignolo¹, Idriz Durmish¹, Barbara Bangol¹, Oliver Wachter¹, Kontanze Hörtnagel¹
¹MVZ Martinsried GmbH, Germany
Introduction
Diagnostics for repeat expansion diseases typically utilize fragment length analysis or Southern blot as separate assays for each indication, providing only limited information on interruptions, methylation status, or mosaics. Our objective is to streamline the laboratory workflow for diagnostics of various repeat diseases, while enhancing detailed characterization of these genomic regions Concurrently, we aim to compare two enrichment methods to assess their effectiveness in this specific application.
Methods
Genomic DNA of 14 samples was isolated from whole blood using Mag Bind Blood Tissue DNA (Omega) and additionally for six of these samples using Puregene Blood Kit (Qiagen). Two Coriell reference samples were also included for analysis.
In these 16 samples, 20 repeat regions were enriched using both a customized hybridization based panel (Twist Bioscience) and the CRISPR Cas 9 method (PureTarget kit, PacBio). Long-read sequencing of all samples was performed on a Sequel IIe (PacBio). Hybridization-enriched samples were sequenced with 15-30h movie time together with other multiplexed amplicon libraries. CRISPR-Cas9-enriched samples were sequenced in a 16-plex setup with 30h movie time, including low quality reads and kinetics information in the analysis output. Data were analyzed using SeqPilot software (JSI) and Tandem Repeat Genotyping Tool (TRGT, PacBio).
Results
CRISPR-Cas9 Enrichment:
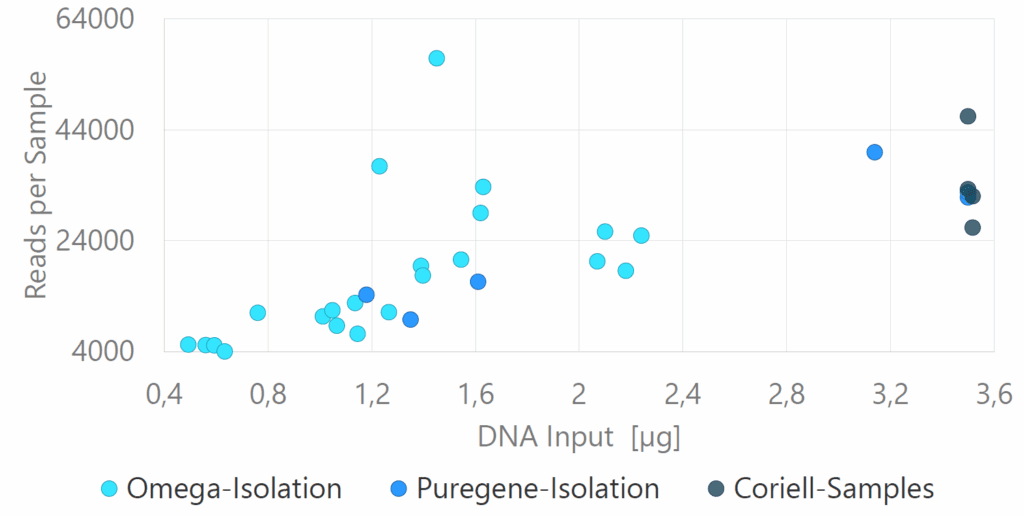
Over two sequencing runs and 16 different samples (15 samples sequenced in both runs), sequencing depth varied between 4,000 and 57,000 reads per sample, depending mainly on DNA input (between 0.5-3.5 µg) and less on the isolation method (Figure 1).
Figure 1. Corrleation between DNA-input and coverage per sample.
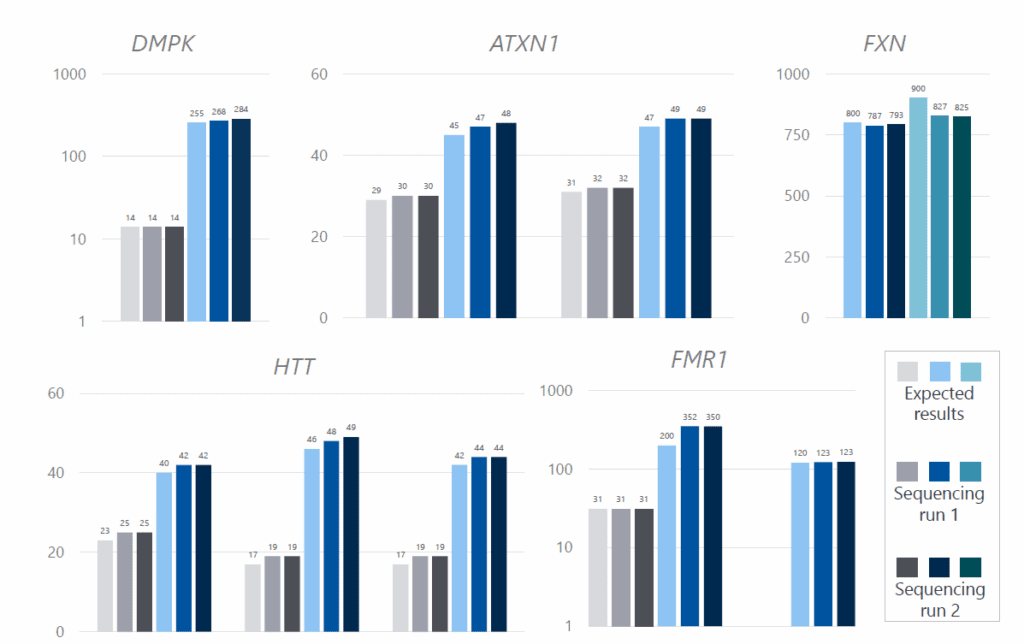
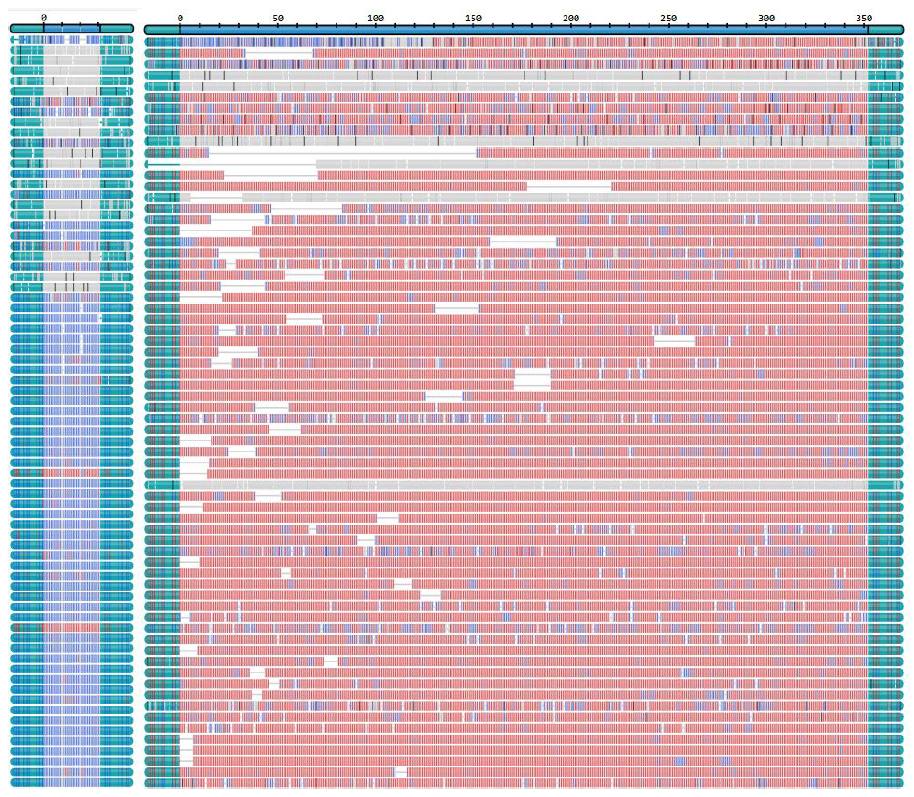
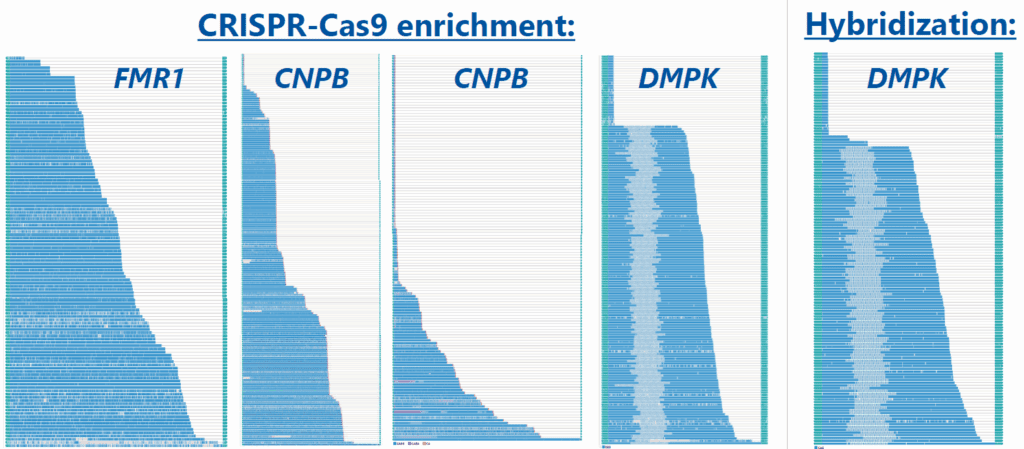
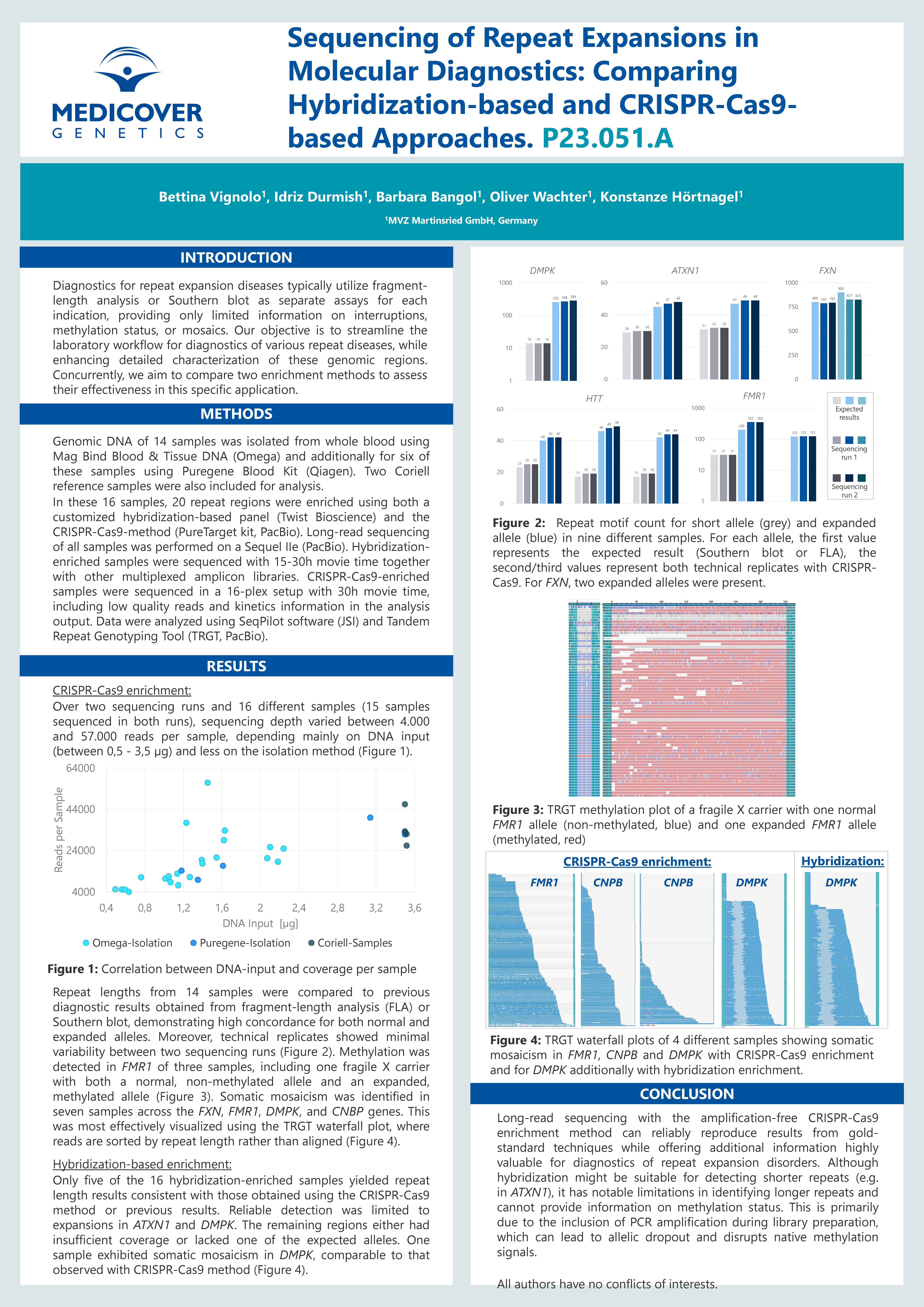
Repeat lengths from 14 samples were compared to previous diagnostic results obtained from fragment length analysis (FLA) or Southern blot, demonstrating high concordance for both normal and expanded alleles. Moreover, technical replicates showed minimal variability between two sequencing runs (Figure 2). Methylation was detected in FMR1 of three samples, including one fragile X carrier with both a normal, non-methylated allele and an expanded, methylated allele (Figure 3). Somatic mosaicism was identified in seven samples across the FXN,FMR1,DMPK, and CNBP genes. This was most effectively visualized using the TRGT waterfall plot, where reads are sorted by repeat length rather than aligned (Figure 4).
Hybridization-based enrichment:
Only five of the 16 hybridization-enriched samples yielded repeat length results consistent with those obtained using the CRISPR-Cas9 method or previous results. Reliable detection was limited to expansions in ATXN1 and DMPK. The remaining regions either had insufficient coverage or lacked one of the expected alleles. One sample exhibited somatic mosaicism in DMPK, comparable to that observed with CRISPR-Cas9 method (Figure 4).
Figure 2. Repeat motif count for short allele (grey) and expanded allele (blue) in nine different samples. For each allele, the first value represents the expected result (Southern blot or FLA), the second/third values represent both technical replicates with CRISPR-Cas9. For FXN, two expanded alleles were present.
Figure 3. TRGT methylation plot of fragile X carrier with one normal FMR1 allele (non-methylated, blue) and one expanded FMR1 allele (methylated, red).
Figure 4. TRGT waterfall plots of 4 different samples showing somatic mosaicism in FMR1, CNPB, and DMPK with CRISPR-Cas9 enrichment and for DMPK additionally with hybridization enrichment.
Conclusion
Long-read sequencing with the amplification-free CRISPR-Cas9 enrichment method can reliably reproduce results from gold-standard techniques while offering additional information highly valuable for diagnostics of repeat expansion disorders. Although hybridization might be suitable for detecting shorter repeats (e.g. in ATXN1), it has notable limitations in identifying longer repeats and cannot provide information on methylation status. This is primarily due to the inclusion of PCR amplification during library preparation, which can lead to allelic dropout and disrupts native methylation signals.
We use cookies to ensure that we give you the best experience on our website. If you continue to use this site we will assume that you are happy with it.